

Diabetes mellitus tip 2 je hronična, progresivna bolest na čiji nastanak utiče više patofizioloških mehanizama. Nastaje kao posljedica različitih abnormalnosti na nivou perfernih tkiva, a karakteriše je povišena koncentracija glukoze u krvi.
Metabolički poremećaji kod DM TIP2 uključuju:
- rezistenciju na inzulinsku aktivnost u jetri, mišićnom i masnom tkivu
- povećanu hepatičnu glukoneogenezu i glukogenolizu
- oštećenu inzulinsku sekreciju
Genetski faktori udruženi sa faktorima okoline dovode do klinički manifestne šećerne bolesti.
Inzulinska rezistencija i defekt u sekreciji inzulina prethode razvoju postprandijalne hiperglikemije i dijabetesa tipa 2.
Tip 2 čini 90% svih slučajeva dijabetesa u svijetu.
Pacijenti koji obolijevaju od ovog oblika DM su obično stariji od 40 godina (najveća incidenca od 65-74 godina) i najveći broj njih je gojazan. Inzulin im nije potreban za preživljavanje na početku bolesti, ali vremenom, zbog iscrpljivanja ß ćelija gušterače, trebaju inzulin radi postizanja optimalne glikoregulacije.
Loše prehrambene navike, politički, ekonomski i socijalni pritisci uz razlike u broju unesenih i potrošenih kalorija najčešće industrijski obrađene hrane, utiču na sve veći broj mladih koji obolijevaju od ovog tipa dijabetesa.
Prava epidemija gojaznosti koja je prisutna širom svijeta posebno u visoko razvijenim industrijskim zemljama, a sve više i u zemljama u razvoju, jedan su od najznačajnijih faktora nastanka DM tipa 2.
Smanjena osjetljivost na inzulin prisutna je kod najvećeg broja oboljelih sa tipom 2 i povezuje se sa: nedefinisanim genetskim faktorima, načinom života, abdominalnom visceralnom gojaznošću.

Oko 30% oboljelih nisu gojazni. Kod takvih je u prvom planu smanjena sekrecija inzulina ali, također i inzulinska rezistencija na postreceptorskom nivou.
TIP2 DM negojaznih osoba u osnovi ima autoimunu destrukciju ß ćelija pankreasa.
Sam tok autoimunog procesa je spor, bolest se obično manifestira nakon 35. godine, a simptomi su nejasni ili blago izraženi. Poslije nekoliko mjeseci ili godina oboljeli trebaju egzogeni inzulin.
Autoimuni inzulitis negojaznih uslovljen je:
1. nasljednom sklonošću (postojanje određene genetske konstitucije koja uključuje ekspresiju HLA DR 3 i 4 HLA A 1,2 i B 8 i 15)
2. još uvijek nepoznatim faktorom, okidačem autoimunog procesa (toksin, virus, abnormalna sekrecija citokina IL 1 ß)
Ovi okidački mehanizmi oslobađaju skrivene proteine iz ß ćelija koje imuni sistem detektuje kao strani, pa odgovor usmjeravaju ka tim proteinima i ß ćelijama što vremenom dovodi do njihove destrukcije.
U osnovi patogeneze tipa 2 DM koji se javlja kod gojaznih osoba je nasljedno uslovljena rezistencija na inzulin. To je stanje u kojem normalna količina inzulina proizvodi oslabljen biološki odgovor na ciljnim ćelijama što ima za posljedicu kompenzatornu hiperinzulinemiju. 70% oboljelih odlikuje visceralna pretilost. Smatra se da visceralne nakupine masnog tkiva imaju veću metaboličku aktivnost od subkutanih nakupina masnog tkiva, pa učinci visceralne pretilosti podrazumijevaju: hiperglikemiju, povećanu količinu SMK u krvi, povećan tonus simpatikusa, oslabljeno inzulinsko signaliziranje, hroničnu upalu, oštećenje endotela i razvoj ateroskleroze.
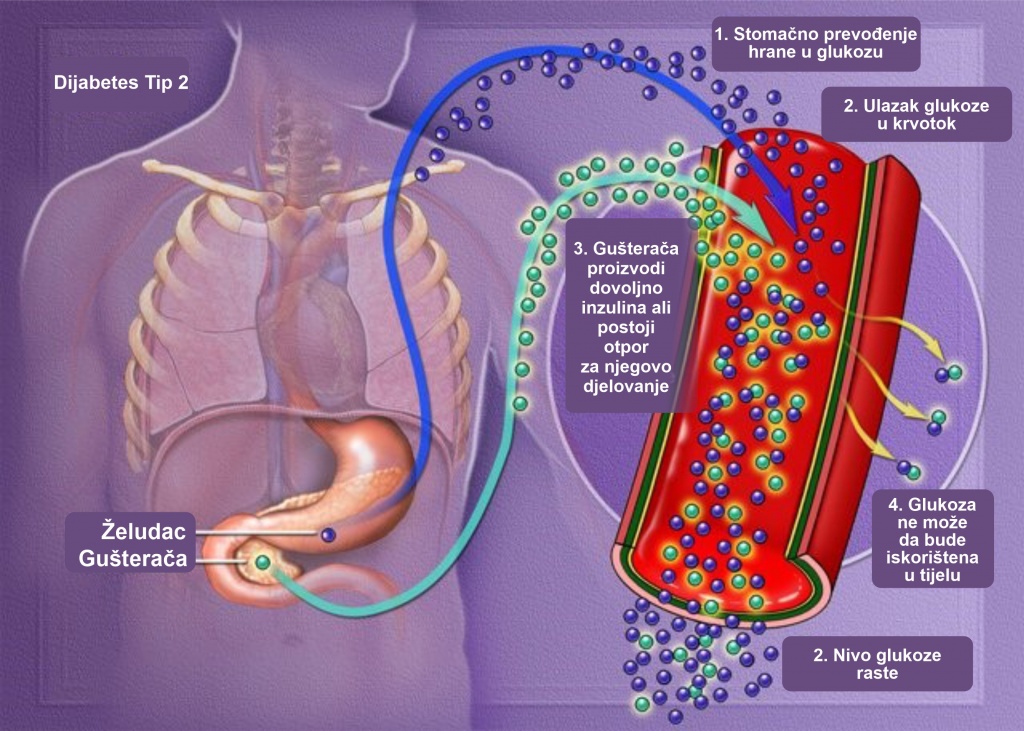
Inzulinska rezistencija (IR) je obično najranija manifestacija u razvoju tipa 2 dijabetesa i nastaje 5 do 10 godina prije pojave postprandijalne hiperglikemije i tipa 2 DM.
Endogeni inzulin koji se luči pri inzulinskoj rezistenciji je nedovoljno aktivan da suprimira hepatičku glukoneogenezu ili stimulira utilizaciju glukoze u mišićnom i masnom tkivu.
Smatra se da abdominalno masno tkivo doprinosi nastanku inzulinske rezistencije tako što otpušta povećane količine faktor tumorske nekroze i slobodne masne kiseline.
Tako se ometa inzulinsko signaliziranje, smanjuje preuzimanje glukoze u mišićima, povećava sinteza triglicerida i potiče glukoneogeneza u jetri.
Inzulinska rezistencija može biti :
1. prereceptorska (postoje antitijela na inzulin koja sprječavaju vezivanje inzulina za receptor)
2. receptorska (mutacije inzulinskih receptora, antitijela na receptorima)
3. postreceptorska (za djelovanje inzulina potrebna je fosforilacija tirozin kinaze u receptoru)
Klinički znaci Inzulinske rezistencije:
1. Povišen inzulin natašte
2. Povišena inzulinska rezistencija (HOMA=IR)
3. Smanjena inzulinska osjetljivost (HOMA=B%)
Inzulinska rezistencija je najčešće metabolička, a rjeđi oblici su sa prisutnim antitijelima na inzulinske receptore.
Rezistencija na inzulin, sa posljedičnom hiperinzulinemijom može trajati godinama i pogoduje razvoju ateroskleroze. Vremenom dolazi do amiliske degeneracije pankreasa, iscrpljivanja ß ćelija, pa osoba iz faze hiperinzulinemije ulazi u fazu hipoinzulinemije.
Rezerve inzulina više nisu dovoljne za prevazilaženje inzulinske rezistencije.
Posljedica:
- Relativni nedostatak inzulina
- Manifestna hiperglikemija
U prvoj fazi je postprandijalna umjerena hiperglikemija naročito nakon obroka bogatih ugljikohidratima (intolerancija glukoze). Druga faza je stalna hiperglikemija tj. i prije obroka .
Na progresiju hiperglikemije utiče i tzv. glikozna toksičnost.
Povećanjem glikemije iznad 7 mmol naglo opada sekrecija inzulina iz ß ćelija pa se taj efekat visokih vrijednosti glikemije naziva glikozna toksičnost.
- Jenkins AB, Campbell LV. The genesis genetics and patophyology of diabetes mellitus. J Inherit Melab Dis 2004; 27: 331-47
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet 2005;365:1415-28.
- Granberry MC, Fonseca VA. Insulin resistance syndrome: options for treatment. South Med J 1999;92:2-15.
- Greenfield JR, Campbell LV. Insulin resistance and obesity. Clin Dermatol 2004;22:289-95.
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214-32.
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412-9.
- Cummings DE, Schwartz MW. Genetics and pathophysiology of human obesity. Annu Rev Med 2003;54:453-71.
- Grundy SM, Brewer HB Jr, Cleeman JI, Smith SC Jr, Lenfant C; American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association Conference on Scientific Issues Related to Definition. Circulation 2004;109:433-8.
- Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004;89:2548-56.
- Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev 200;14:447-55.
- Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 1995;95:2111-9.

